PXR-dependent induction of SREBP1a largely determines induction of lipogenic SREBP1 target genes
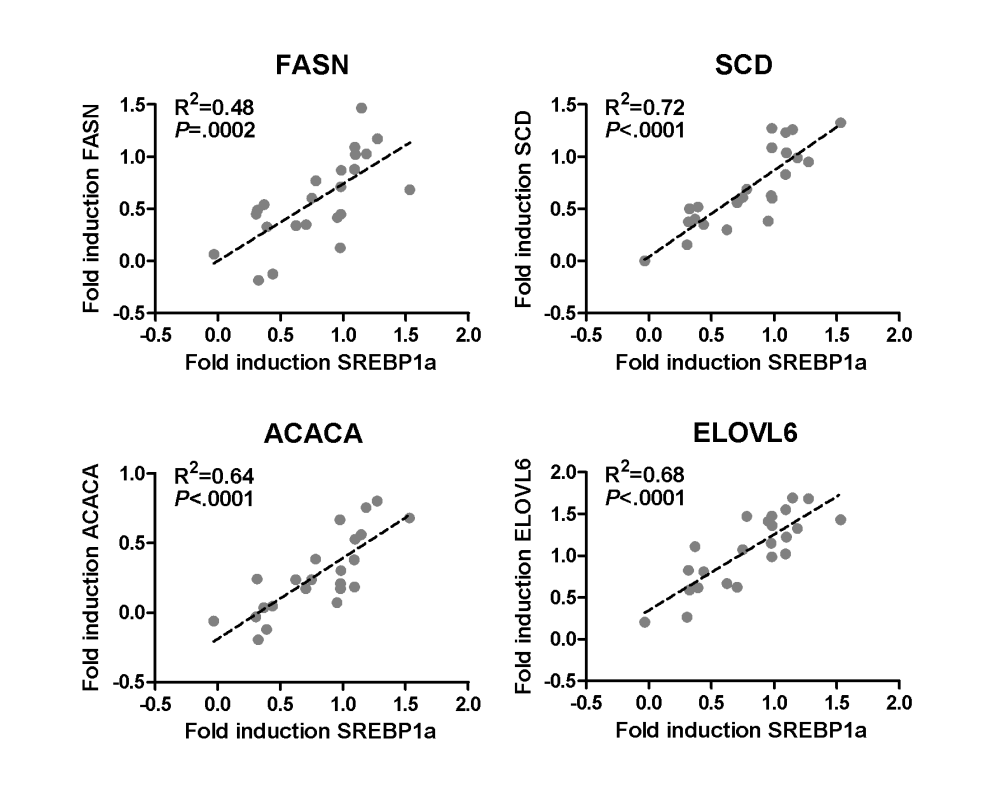 Primary human hepatocyte cultures of 23 donors were treated with 30 μM rifampicin for 48 h. Linear regression analysis of log2-transformed values of fold induction by rifampicin of the mRNA expression of SREBP1a and the indicated genes in primary human hepatocytes, as shown in Fig. 3a in the article »Pregnane X receptor activation and silencing promote steatosis of human hepatic cells by distinct lipogenic mechanisms« (see Suppl. material).
Primary human hepatocyte cultures of 23 donors were treated with 30 μM rifampicin for 48 h. Linear regression analysis of log2-transformed values of fold induction by rifampicin of the mRNA expression of SREBP1a and the indicated genes in primary human hepatocytes, as shown in Fig. 3a in the article »Pregnane X receptor activation and silencing promote steatosis of human hepatic cells by distinct lipogenic mechanisms« (see Suppl. material).
Details about the proposed molecular mechanisms of PXR-dependent induction of SREBP1a can be found below the Animation Scheme
First mechanism
1. Both LSD1 and PXR interact with SIRT1
2. LSD1 increases transcription (maybe through demethylation of Sp1) and DNA binding affinity of SREBP1a
3. SREBP1a is acetylated by p300 and deacetylated by SIRT1
A: Giandomenico et al. Coactivator-dependent acetylation stabilizes members of the SREBP family of transcription factors. Mol Cell Biol. 2003 Apr;23(7):2587-99.
Second mechanism
1. PXR induces p38 MAPK phosphorylation
2. Relocation of hnRNP A1 and the activation of SREBP1a are dependent on the p38 MAPK signal pathway
A crucial point to note:
SIRT1 is an important hub interacting with a complex network of proteins. However, there are plausible reasons to assume that the interaction of PXR with SIRT1 not only activates SREBP1a but also influences total body energy metabolism (see animation below) .
Furthermore, I suppose that PXR not only activates SREBP1a but also represses the transcription of SREBP1c via SIRT1 in a LXR-dependent manner (SREBP1a-dependent induction of 1c remains!):
A: Li et al. SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol Cell. 2007 Oct 12;28(1):91-106.
B: Bitter et al. Pregnane X receptor activation and silencing promote steatosis of human hepatic cells by distinct lipogenic mechanisms. Arch Toxicol. 2015 Nov;89(11):2089-103. [See figure 3a and supplementary figure S4]
One important question remains:
Which ligand (or combination of ligands!) could be responsible for an elevated activation of the pregnane X receptor PXR?
Finally, the ligands must have properties to activate PXR chronically and deliver the performance to activate PXR in an additive or Synergistic Way
And in this context, the following proposed mechanisms (and the combination of both!) seem to be highly relevant:
1. Exogenous PXR ligands
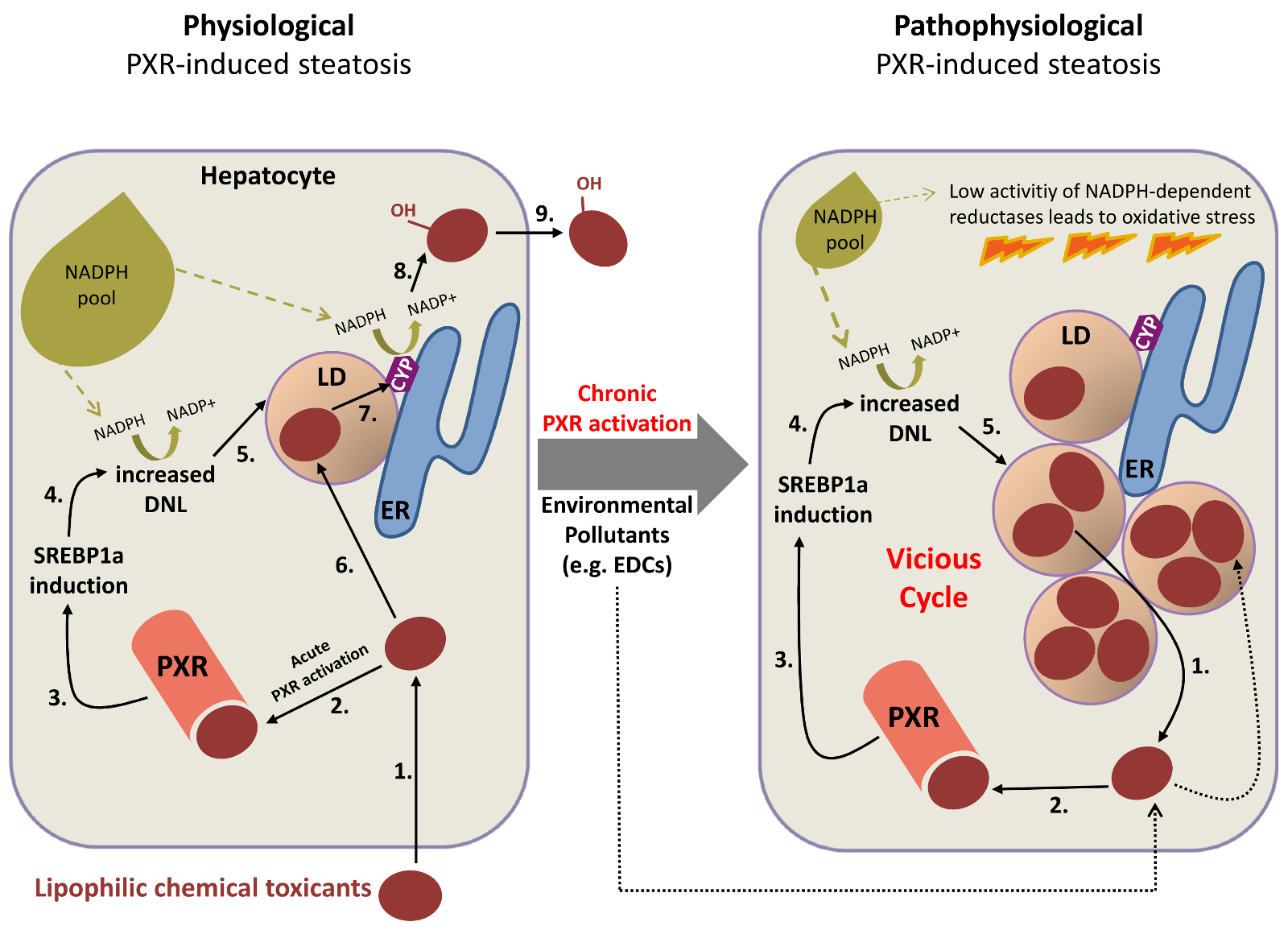 1. Exogenous PXR ligands: Lipophilic chemical toxicants activate PXR. DNL, de novo lipogenesis; LD, lipid droplet; ER, endoplasmic reticulum; CYP, cytochrome P450; EDC, endocrine disrupting chemical. Numbers indicate the proposed sequence of events.
1. Exogenous PXR ligands: Lipophilic chemical toxicants activate PXR. DNL, de novo lipogenesis; LD, lipid droplet; ER, endoplasmic reticulum; CYP, cytochrome P450; EDC, endocrine disrupting chemical. Numbers indicate the proposed sequence of events.
***
The central role of storing chemical toxicants in lipid droplets for the physiology and pathophysiology of pregnane X receptor-induced human hepatocellular steatosis
Hypothesis
To explain the apparent contradiction of the parallel induction of detoxification and de novo lipogenesis, in terms of competition for the NADPH pool, it can be postulated that the steatogenic effect of acute PXR activation is a protective physiological mechanism to provide more buffer capacity/safe storage locations for potentially toxic compounds in the induced lipid droplets of hepatocytes and to promote regulated detoxification by the cytochrome P450 enzyme system (Upper figure, left part). This enzyme system is localized in the endoplasmatic reticulum, to which lipid droplets are closely associated or even connected. The hypothesis newly ascribes a physiological function to de novo lipogenesis and lipid droplet formation due to the acute activation of PXR, which may also be the cause of the pathophysiological situation potentially arising in chronic PXR activation. If PXR activation is maintained for long by continuous exposure to e.g. environmental pollutants being PXR ligands, this is suggested to result in lipid droplets steadily growing both in size and numbers, thereby leading to progressive accumulation of lipophilic toxic chemicals in the lipid droplets. A vicious cycle of further PXR activation, increasing de novo lipogenesis/steatosis and accumulation of PXR activating toxic lipophilic chemicals is thus predicted (Upper figure, right part). Finally, this should result in oxidative stress by exhausting NADPH pools, which are essential for e.g. the enzymatic activity of the glutathione reductase, and consequent cellular damage.
The concept is solely based on theoretical considerations and thus contains a certain risk of failure. However, it is completely new and groundbreaking and if true will not only provide an explanation for the starting observation, but will also open new views on detoxification as well as on the etiology of NAFLD/NASH.
2. Endogenous PXR ligands
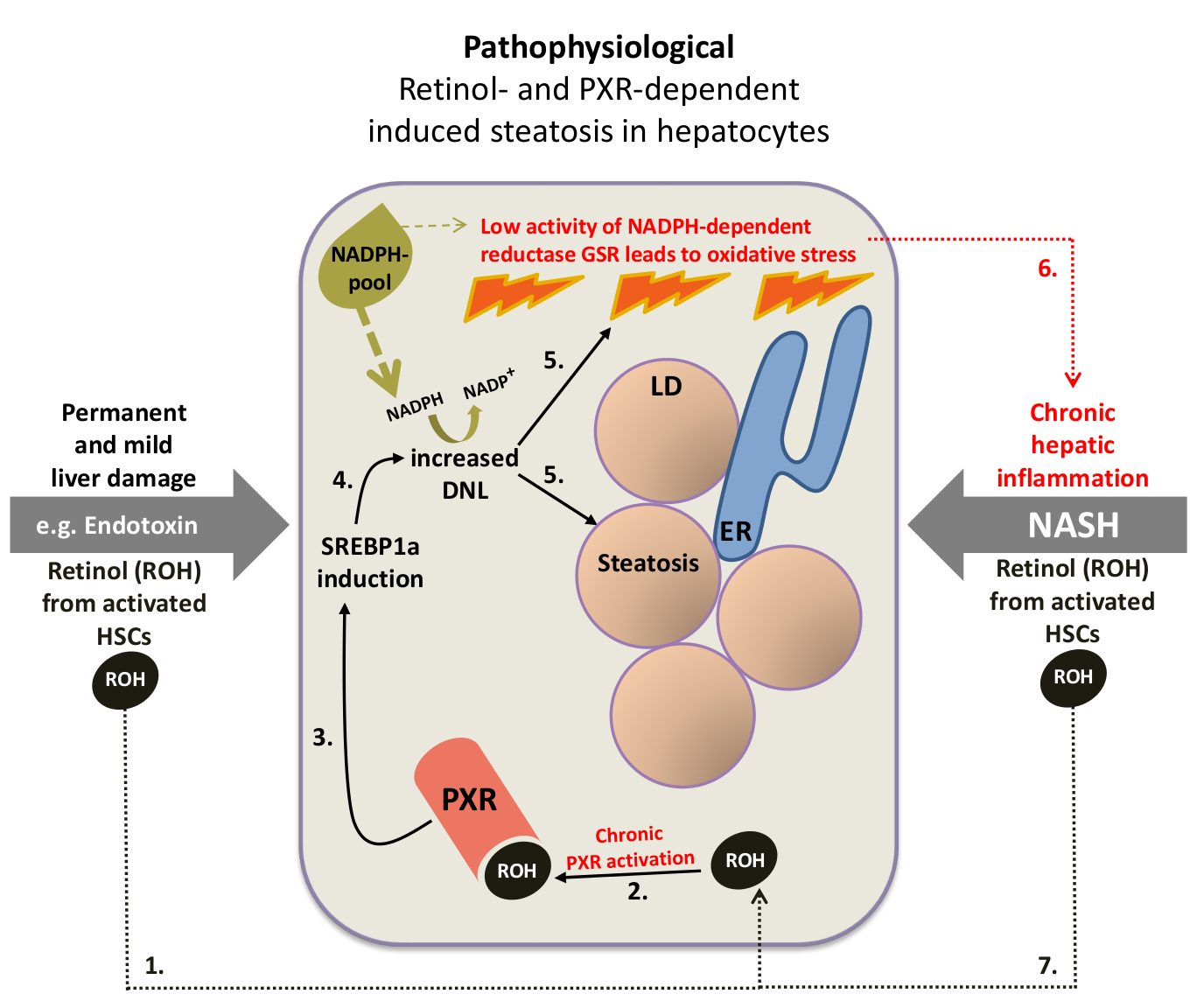 2. Endogenous PXR ligands: Retinol activates PXR. DNL, de novo lipogenesis; LD, lipid droplet; ER, endoplasmic reticulum; HSC, hepatic stellate cell; GSR, glutathione reductase. Numbers indicate the proposed sequence of events.
2. Endogenous PXR ligands: Retinol activates PXR. DNL, de novo lipogenesis; LD, lipid droplet; ER, endoplasmic reticulum; HSC, hepatic stellate cell; GSR, glutathione reductase. Numbers indicate the proposed sequence of events.
One Example for Future Research
Already in 2012, I received the opportunity to discuss about my work and hypotheses in form of presentations:
(May 20 – 22) 1st IKP Stuttgart Scientific Retreat: The link between intestinal microbiota, hepatic PXR and NAFLD.
(February 29 – March 1) EASL Basic School of Hepatology Course 7: Hepatocyte Damage & Liver Metabolism: The role of PXR in human hepatic steatosis: Establishment of a cellular model.
If you are interested in the full version of the possible future research project presented below, please contact me (non-profit-oriented).
***
The pathophysiological role of the PXR-dependent elevation of de novo lipogenesis in non-alcoholic steatohepatitis (NASH)
Abstract
Non-alcoholic steatohepatitis (NASH), the chronic inflammation of the liver with concomitant fatty liver (steatosis), represents, besides alcohol abuse and hepatic viral infections, the leading risk factor for developing liver cancer. Hepatic oxidative stress is regarded as the key component in the pathogenesis of NASH. Furthermore, recent investigations have shown that the pathological increase of newly synthesized fatty acids in liver (hepatic de novo lipogenesis) represents a pathophysiological process, which contributes in a decisive manner to the development of steatosis and the progression of non-alcoholic fatty liver disease (NAFLD). However, the molecular mechanisms in the pathogenesis and progression of NASH are still remain largely unknown. My recently published work, concerning the role of the pregnane X receptor PXR and SREBP1a in human hepatic de novo lipogenesis and NASH, suggests that both PXR and its target gene SREBP1, the master transcriptional regulator of fatty acid synthesis, constitute a key function in the development and progression of NASH. I propose that the cause of the permanent and significantly increased hepatic de novo lipogenesis in NASH patients is based on the combination of PXR-deficiency and a retinol-dependent increased ligand-induced activity of the still existing PXR. In addition to the investigation of the role of PXR in the retinol-mediated interaction of hepatic stellate cells (HSCs) and hepatocytes, the main attention shall lie on the analysis about the pathophysiologically relevant consequence of a PXR-mediated chronically elevated de novo lipogenesis, the origin of oxidative stress. The resulting findings will contribute to a better understanding of the role of PXR and SREBP1a in NASH and may help devising new therapeutic approaches.
Look inside (~45.000 characters and 5 figures)